The autonomic nervous system (ANS) is essential for survival and responsible for the body’s involuntary activities such as cardiovascular, gastrointestinal, and thermoregulatory homeostasis. The ANS is divided into two major branches: the sympathetic nervous system (SNS), which controls the “fight or flight” responses, and the parasympathetic nervous system (PNS), which oversees the body’s maintenance functions including digestion. Both disease states and the stress of surgery can lead to changes in the ANS that can have potentially deleterious effects. Thus, a primary goal of anesthetic management is to modulate the body’s autonomic responses. Contemporary anesthesia providers have access to many pharmacologic drugs that can profoundly alter autonomic activity; thus, a thorough understanding of the anatomy and physiology of the ANS is essential.
1. ANATOMY OF THE AUTONOMIC NERVOUS SYSTEM
The Sympathetic Nervous System
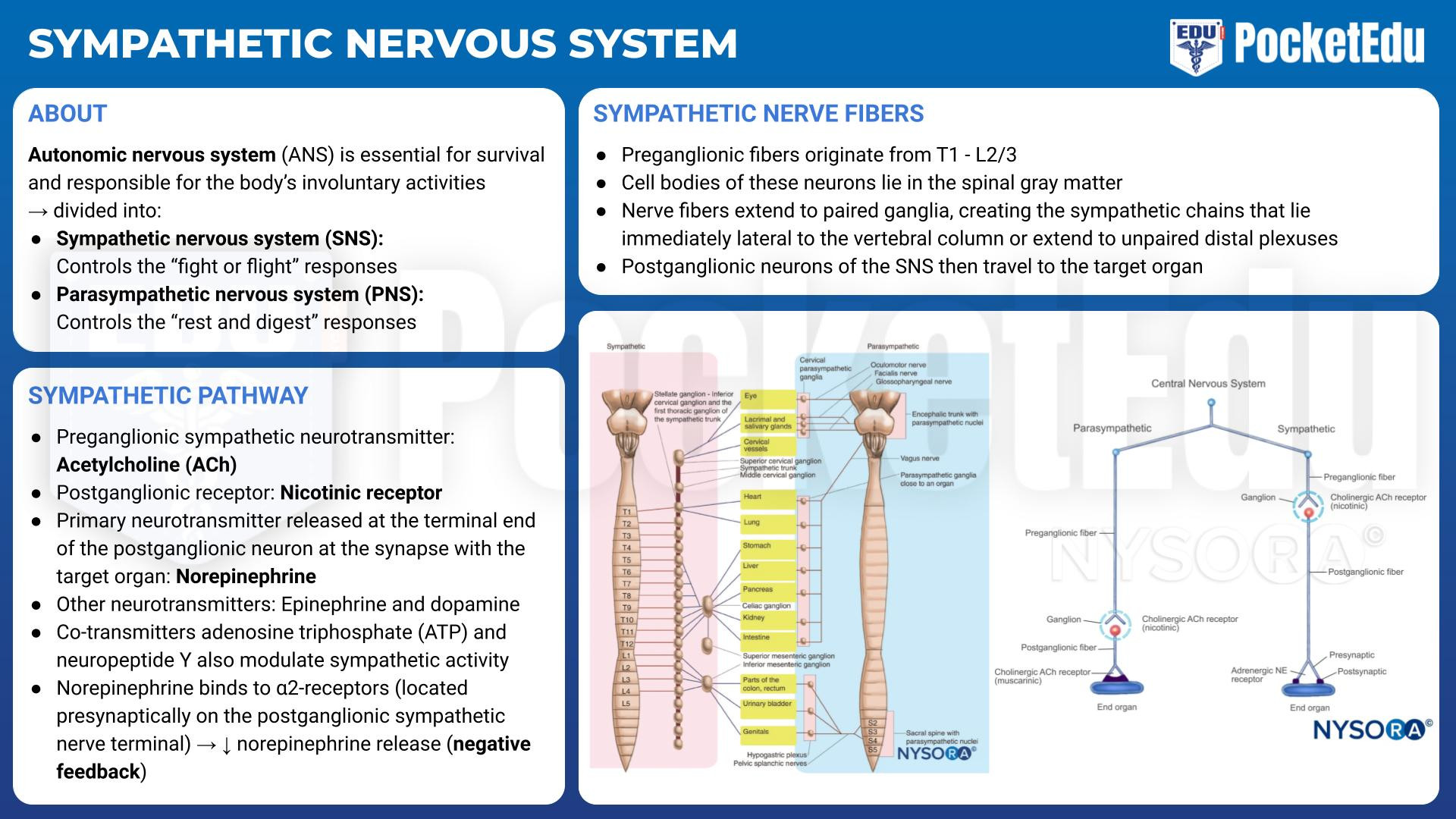
- PockedtEdu Anesthesia Infographics: Sympathetic nervous system 6.1.
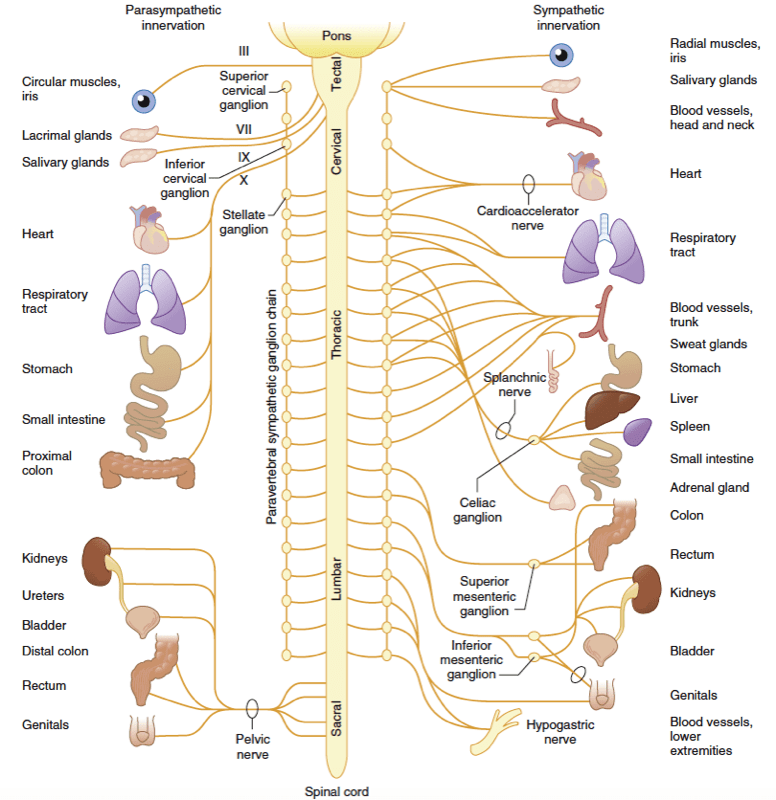
The preganglionic fibers of the SNS originate from the thoracolumbar region (T1 to L2 or L3) of the spinal cord (Fig.1). The cell bodies of these neurons lie in the spinal gray matter. The nerve fibers extend to paired ganglia, creating the sympathetic chains that lie immediately lateral to the vertebral column or extend to unpaired distal plexuses (e.g., the celiac and mesenteric plexuses). Preganglionic sympathetic fibers not only synapse at the ganglion of the level of their origin in the spinal cord but can also course up and down the paired ganglia. A sympathetic response, therefore, is not confined to the segment from which the stimulus originates, as discharge can be amplified and diffuse. The postganglionic neurons of the SNS then travel to the target organ. The sympathetic preganglionic fibers are relatively short because sympathetic ganglia are generally close to the central nervous system (CNS). In contrast, the postganglionic fibers run a long course before innervating effector organs (Fig.2).

- Fig 1. Schematic representation of the autonomic nervous system depicting the functional innervation of peripheral effector organs and the anatomic origin of peripheral autonomic nerves from the spinal cord. Although both paravertebral sympathetic ganglia chains are presented, the sympathetic innervation to the peripheral effector organs is shown only on the right side of the figure, whereas the parasympathetic innervation of peripheral effector organs is depicted on the left. The roman numerals on nerves originating in the tectal region of the brainstem refer to the cranial nerves that provide parasympathetic outflow to the effector organs of the head, neck, and trunk. (From Ruffolo R. Physiology and biochemistry of the peripheral autonomic nervous system. In Wingard L, Brody T, Larner J, et al, eds. Human Pharmacology: Molecular to Clinical. St. Louis: Mosby-Year Book; 1991:77.)

- Fig. 2 Schematic diagram of the peripheral autonomic nervous system. Preganglionic fibers and postganglionic fibers of the parasympathetic nervous system release acetylcholine (ACh) as the neurotransmitter. Postganglionic fibers of the sympathetic nervous system release norepinephrine (NE) as the neurotransmitter (exceptions are fibers to sweat glands, which release ACh). (From Lawson NW, Wallfisch HK. Cardiovascular pharmacology: a new look at the pressors. In Stoelting RK, Barash J, eds. Advances in Anesthesia. Chicago: Year Book Medical Publishers; 1986:195-270.)
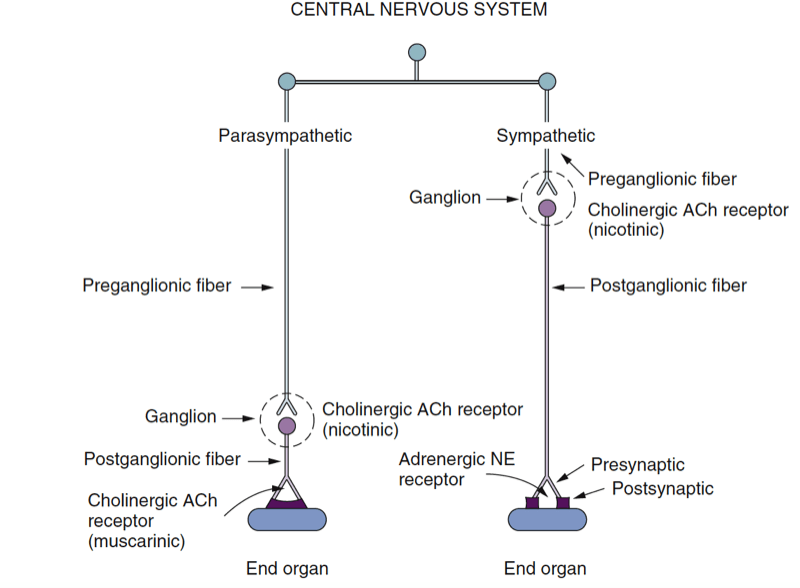
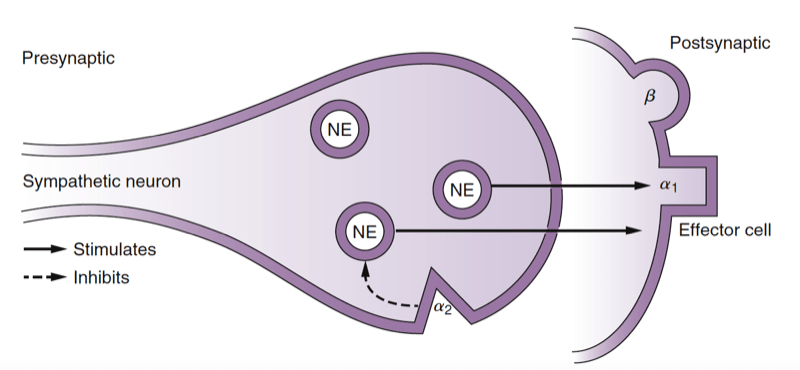
The neurotransmitter released at the terminal end of the preganglionic sympathetic neuron is acetylcholine (ACh), and the cholinergic receptor on the postganglionic neuron is a nicotinic receptor. Norepinephrine is the primary neurotransmitter released at the terminal end of the postganglionic neuron at the synapse with the target organ (Fig.3). Other classic neurotransmitters of the SNS include epinephrine and dopamine. Additionally, co-transmitters, such as adenosine triphosphate (ATP) and neuropeptide Y, modulate sympathetic activity. Norepinephrine and epinephrine bind postsynaptically to adrenergic receptors, which include the α1-, β1-, β2-, and β3-receptors. When norepinephrine binds to the α2- receptors, located presynaptically on the postganglionic sympathetic nerve terminal, subsequent norepinephrine release is decreased (negative feedback). Dopamine (D) binds to D1 receptors postsynaptically or D2 receptors presynaptically.

- Fig 3. Schematic depiction of the postganglionic sympathetic nerve ending. Release of the neurotransmitter norepinephrine (NE) from the nerve ending results in stimulation of postsynaptic receptors, which are classified as α1, β1, and β2. Stimulation of presynaptic α2-receptors results in inhibition of NE release from the nerve ending. (Adapted from Ram CVS, Kaplan NM. Alpha- and beta-receptor blocking drugs in the treatment of hypertension. In Harvey WP, ed. Current Problems in Cardiology. Chicago: Year Book Medical Publishers; 1970.)
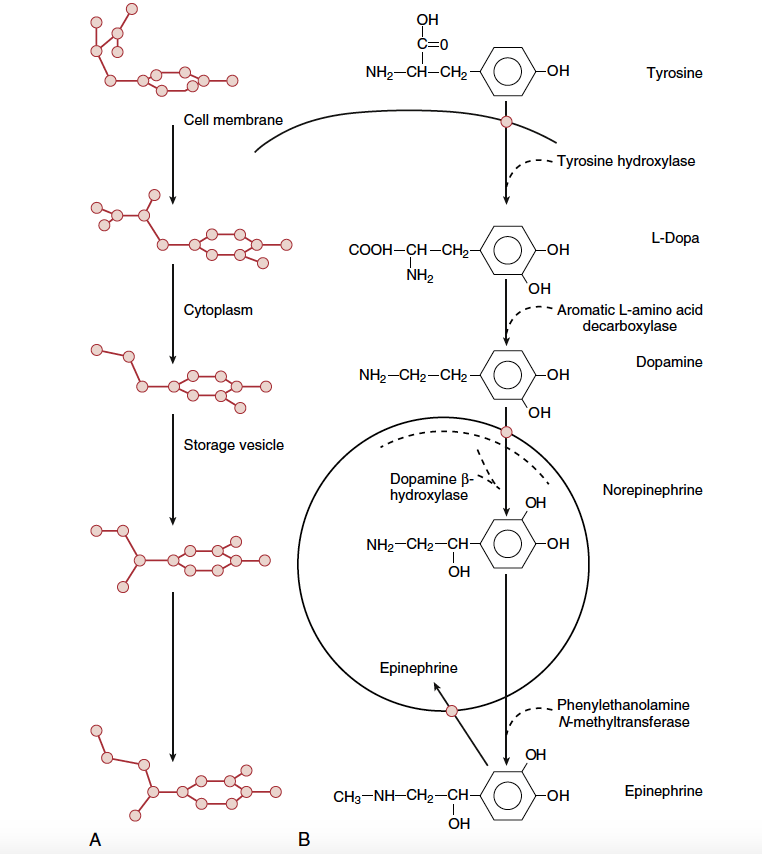
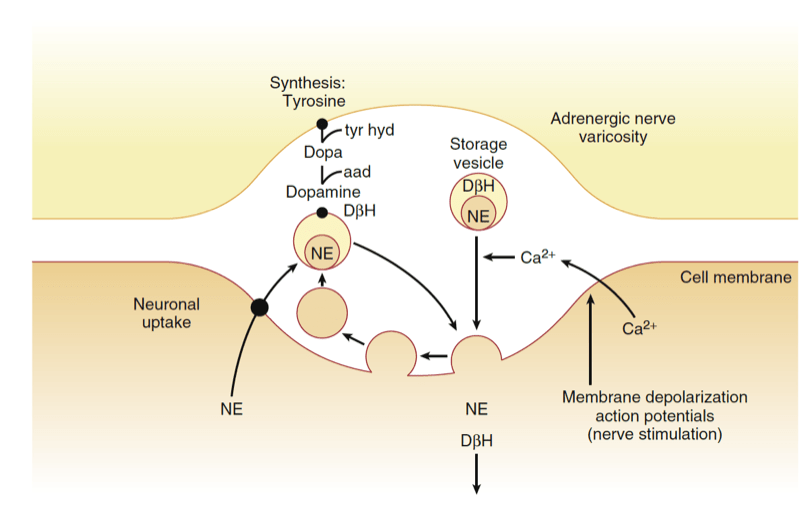
Sympathetic neurotransmitters are synthesized from tyrosine in the postganglionic sympathetic nerve ending (Fig.4). The rate-limiting step is the transformation of tyrosine to dihydroxyphenylalanine (DOPA), which is catalyzed by the enzyme tyrosine hydroxylase. DOPA is then converted to dopamine and, once inside the storage vesicle at the nerve terminal, is β-hydroxylated to norepinephrine. In the adrenal medulla, norepinephrine is methylated to epinephrine. The neurotransmitters are stored in vesicles until the postganglionic nerve is stimulated. Then the vesicles merge with the cell membrane and release their contents into the synapse (Fig.5). In general, only 1% of the total stored norepinephrine is released with each depolarization; thus, there is a tremendous functional reserve. The released norepinephrine binds to the pre- and postsynaptic adrenergic receptors. The postsynaptic receptors then activate secondary messenger systems in the postsynaptic cell via G protein–linked activity. Norepinephrine is then released from these receptors and mostly taken up at the presynaptic nerve terminal and transported to storage vesicles for reuse. Norepinephrine that escapes this reuptake process and makes its way into the circulation is metabolized by either the monoamine oxidase (MAO) or catechol- O-methyltransferase (COMT) enzyme in the blood, liver, or kidney.

- Fig 4. Biosynthesis of norepinephrine and epinephrine in sympathetic nerve terminal (and adrenal medulla). (A) Perspective view of molecules. (B) Enzymatic processes. (From Tollenaer. JP. Atlas of the Three-Dimensional Structure of Drugs. Amsterdam: Elsevier North-Holland; 1979, as modified by Vanhoutte PM. Adrenergic neuroeffector interaction in the blood vessel wall. Fed Proc. 1978;37:181.)

- Fig.5 Release and reuptake of norepinephrine at sympathetic nerve terminals. Solid circle, Active carrier; aad, aromatic l-amino acid decarboxylase; DβH, dopamine β-hydroxylase; Dopa, l-dihydroxyphenyalanine; NE, norepinephrine; tyr hyd, tyrosine hydroxylase. (From Vanhoutte PM. Adrenergic neuroeffector interaction in the blood vessel wall. Fed Proc. 1978;37:181, as modified by Shepherd J, Vanhoutte P. Neurohumoral regulation. In Shepherd S, Vanhoutte P, eds. The Human Cardiovascular System: Facts and Concepts. New York: Raven Press; 1979:107.)
The Parasympathetic Nervous System

- PockedtEdu Anesthesia Infographics: Parasympathetic nervous system 6.2.
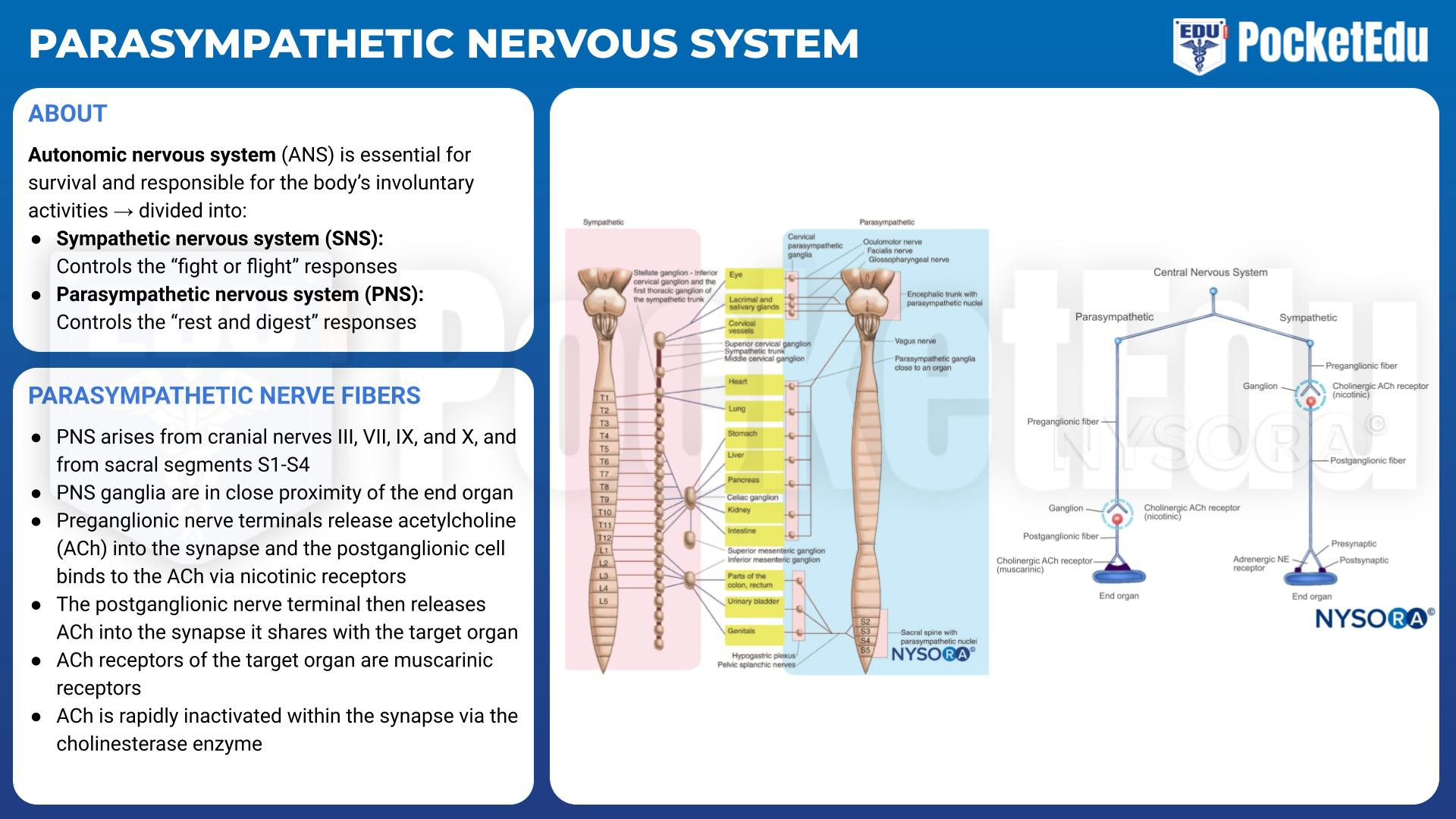
The PNS arises from cranial nerves III, VII, IX, and X as well as from sacral segments S1-S4 (see Fig.1). Unlike the ganglia of the SNS, the ganglia of the PNS are in close proximity to (or even within) their target organs (see Fig.2). Like the SNS, the preganglionic nerve terminals release ACh into the synapse, and the postganglionic cell binds the ACh via nicotinic receptors. The postganglionic nerve terminal then releases ACh into the synapse it shares with the target organ cell. The ACh receptors of the target organ are muscarinic receptors. Like the adrenergic receptors, muscarinic receptors are coupled to G proteins and secondary messenger systems. ACh is rapidly inactivated within the synapse by the cholinesterase enzyme.
2. ADRENERGIC PHARMACOLOGY
Endogenous Catecholamines
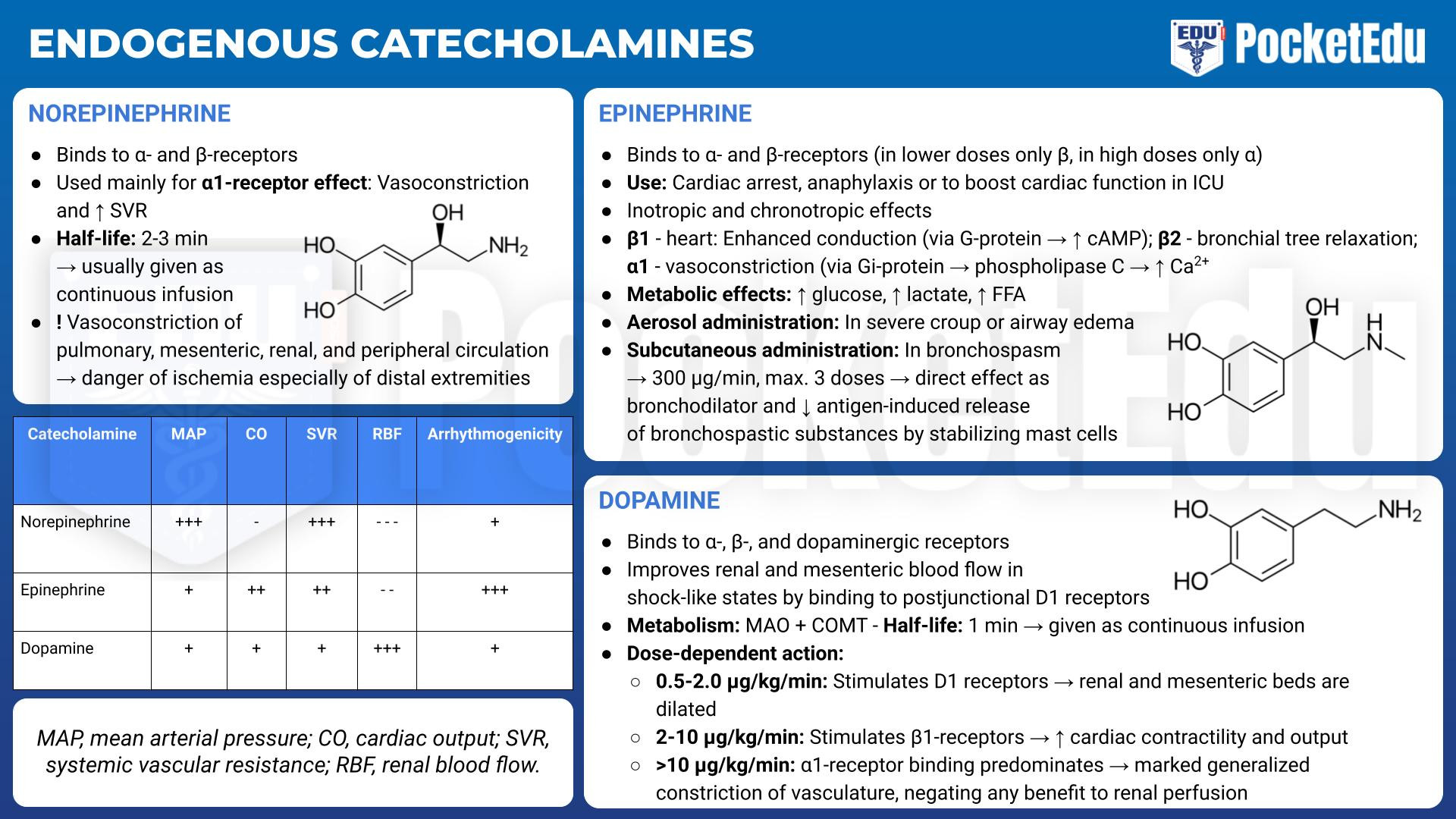
Norepinephrine
Norepinephrine, the primary adrenergic neurotransmitter, binds to α- and β-receptors. It is used primarily for its α1-adrenergic effects that increase systemic vascular resistance. Like all the endogenous catecholamines, the half-life of norepinephrine is short (2.5 minutes), so it is usually given as a continuous infusion at rates of 3 μg/min or more and titrated to the desired effect. The increase in systemic resistance can lead to reflex bradycardia. Additionally, because norepinephrine vasoconstricts the pulmonary, renal, and mesenteric circulations, infusions must be carefully monitored to prevent injury to vital organs. Prolonged infusion of norepinephrine can also cause ischemia in the fingers and toes because of the marked peripheral vasoconstriction.
Epinephrine
Like norepinephrine, epinephrine binds to α- and β- adrenergic receptors. Exogenous epinephrine is used intravenously in life-threatening circumstances to treat cardiac arrest, circulatory collapse, and anaphylaxis. It is also commonly used locally to decrease the systemic absorption of local anesthetics and to reduce surgical blood loss. Among the therapeutic effects of epinephrine are positive inotropy, chronotropy, and enhanced conduction in the heart (β1); smooth muscle relaxation in the vasculature and bronchial tree (β2); and vasoconstriction (α1). The effects that predominate depend on the dose of epinephrine administered. Epinephrine also has endocrine and metabolic effects that include increasing the levels of blood glucose, lactate, and free fatty acids.
An intravenous dose of 1 mg can be given for cardiovascular collapse, asystole, ventricular fibrillation, pulseless electrical activity, or anaphylactic shock to constrict the peripheral vasculature and maintain myocardial and cerebral perfusion. In less acute circumstances, epinephrine can be given as a continuous infusion. The response of individual patients to epinephrine varies, so the infusion must be titrated to effect while the patient is monitored for signs of compromised renal, cerebral, or myocardial perfusion. In general, an infusion rate of 1 to 2 μg/min should primarily stimulate β2-receptors and decrease airway resistance and vascular tone. A rate of 2 to 10 μg/min increases heart rate, contractility, and conduction through the atrioventricular node. When doses larger than 10 μg/min are given, the α1-adrenergic effects predominate, resulting in generalized vasoconstriction, which can lead to reflex bradycardia. Epinephrine also can be administered as an aerosol to treat severe croup or airway edema. Bronchospasm is treated with epinephrine administered subcutaneously in doses of 300 μg every 20 minutes with a maximum of three doses. Epinephrine treats bronchospasm both via its direct effect as a bronchodilator and because it decreases antigen-induced release of bronchospastic substances (as may occur during anaphylaxis) by stabilizing the mast cells that release these substances. Because epinephrine decreases the refractory period of the myocardium, the risk of arrhythmias during halothane anesthesia is increased when epinephrine is given. The risk of arrhythmias seems to be less in children but increases with hypocapnia.
Dopamine
In addition to binding to α- and β-receptors, dopamine binds to dopaminergic receptors. Besides its direct effects, dopamine acts indirectly by stimulating the release of norepinephrine from storage vesicles. Dopamine is unique in its ability to improve blood flow through the renal and mesenteric beds in shock-like states by binding to postjunctional D1 receptors. Dopamine is rapidly metabolized by MAO and COMT and has a half-life of 1 minute, so it must be given as a continuous infusion. At doses between 0.5 and 2.0 μg/ kg/min, D1 receptors are stimulated and renal and mesenteric beds are dilated. When the infusion is increased to 2 to 10 μg/kg/min, the β1-receptors are stimulated and cardiac contractility and output are increased. At doses of 10 μg/kg/min and higher, α1-receptor binding predominates and causes marked generalized constriction of the vasculature, negating any benefit to renal perfusion. In the past, dopamine was frequently used to treat patients in shock. The belief was that infusions of dopamine, by improving renal blood flow, could protect the kidney and aid in diuresis. Subsequently, dopamine was not found to have a beneficial effect on renal function in shock states. Its routine use for patients in shock is questionable because it may increase mortality risk and the incidence of arrhythmic events. (1,2)
Synthetic Catecholamines
- PockedtEdu Anesthesia Infographics: Synthetic catecholamines 6.4.
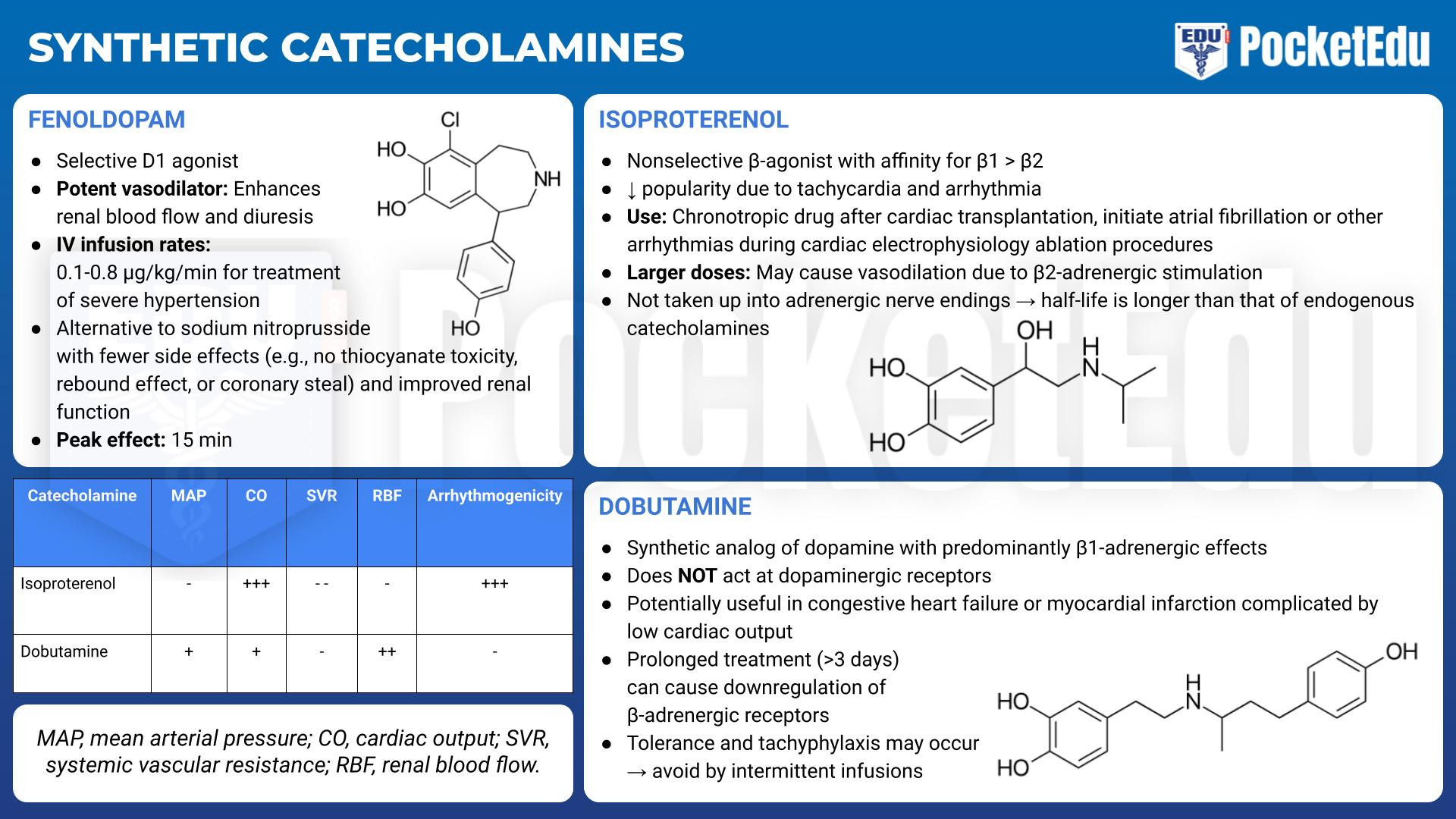
Isoproterenol
Isoproterenol (Isuprel) provides relatively pure and nonselective β-adrenergic stimulation. Its β1-adrenergic stimulation is greater than its β2-adrenergic effects. Its popularity has declined because of adverse effects such as tachycardia and arrhythmias. It is no longer part of the Advanced Cardiac Life Support protocols , and its principal uses now are as a chronotropic drug after cardiac transplantation and to initiate atrial fibrillation or other arrhythmias during cardiac electrophysiology ablation procedures. With larger doses, isoproterenol may cause vasodilation due to β2-adrenergic stimulation. Because isoproterenol is not taken up into the adrenergic nerve endings, its half-life is longer than that of the endogenous catecholamines.
Dobutamine
Dobutamine, a synthetic analog of dopamine, has predominantly β1-adrenergic effects. When compared with isoproterenol, inotropy is more affected than chronotropy. It exerts less of a β2-type effect than isoproterenol does and less of an α1-type effect than does norepinephrine. Unlike dopamine, endogenous norepinephrine is not released, and dobutamine does not act at dopaminergic receptors. Dobutamine is potentially useful in patients with congestive heart failure (CHF) or myocardial infarction complicated by low cardiac output. Doses smaller than 20 μg/kg/min usually do not produce tachycardia. Because dobutamine directly stimulates β1-receptors, it does not rely on endogenous norepinephrine stores for its effects and may still be useful in catecholamine-depleted states such as chronic CHF. However, prolonged treatment with dobutamine causes downregulation of β-adrenergic receptors. If given more than 3 days, tolerance and even tachyphylaxis may occur and can be avoided by intermittent infusions of dobutamine. However, there are no controlled trials demonstrating improved survival. (3)
Fenoldopam
Fenoldopam is a selective D1 agonist and potent vasodilator that enhances renal blood flow and diuresis. Because of mixed results in clinical trials, fenoldopam is no longer used for treatment of chronic hypertension or CHF. Instead, intravenous fenoldopam, at infusion rates of 0.1 to 0.8 μg/kg/min, has been approved for treatment of severe hypertension. Fenoldopam is an alternative to sodium nitroprusside with fewer side effects (e.g., no thiocyanate toxicity, rebound effect, or coronary steal) and improved renal function. Its peak effect takes 15 minutes.
Noncatecholamine Sympathomimetic Amines
- PockedtEdu Anesthesia Infographics: Noncatecholamine sympathomimetics and sympatholytics 6.5.
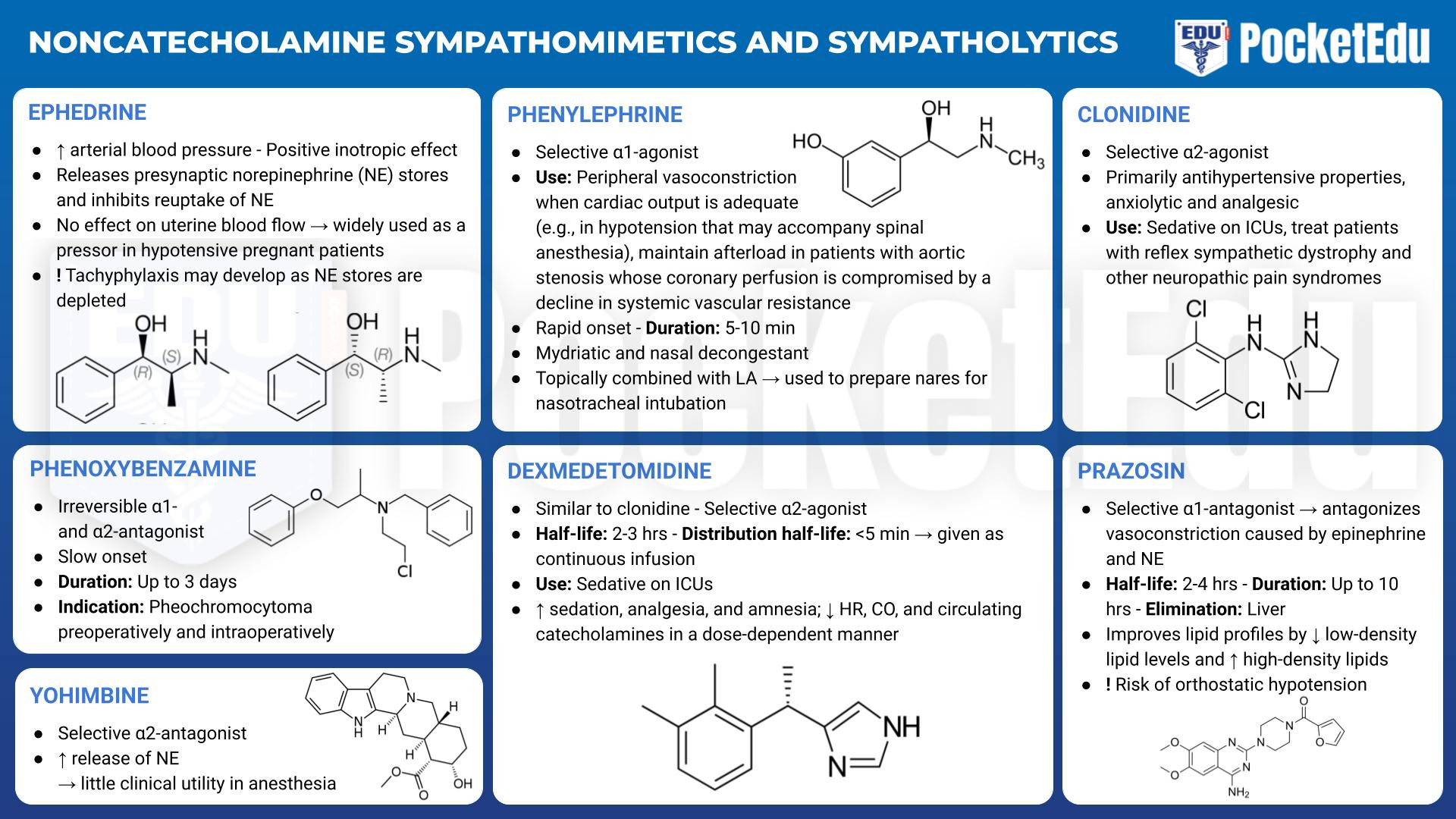
Most noncatecholamine sympathomimetic amines act at α- and β-receptors through both direct (binding of the drug by adrenergic receptors) and indirect (release of endogenous norepinephrine stores) activity. Mephentermine and metaraminol are rarely used currently, so the only widely used noncatecholamine sympathomimetic amine at this time is ephedrine.
Ephedrine
Ephedrine increases arterial blood pressure and has a positive inotropic effect. Because it does not have detrimental effects on uterine blood flow in animal models, ephedrine became widely used as a pressor in hypotensive pregnant patients. However, phenylephrine is now the preferred treatment for hypotension in the parturient because of a decreased risk of fetal acidosis. As a result of its β1-adrenergic stimulating effects, ephedrine is helpful in treating moderate hypotension, particularly if accompanied by bradycardia. The usual dose is 2.5 to 10 mg given intravenously or 25 to 50 mg administered intramuscularly. Tachyphylaxis to the indirect effects of ephedrine may develop as norepinephrine stores are depleted. In addition, although drugs with indirect activity are widely used as a first-line therapy for intraoperative hypotension, repeat doses of ephedrine administration in life-threatening events (instead of switching to epinephrine) may contribute to morbidity. (4)
3. SELECTIVE Α-ADRENERGIC RECEPTOR AGONISTS
α(1)-Adrenergic Agonists
Phenylephrine
Phenylephrine (Neo-Synephrine), a selective α1-agonist, is frequently used for peripheral vasoconstriction when cardiac output is adequate (e.g., in the hypotension that may accompany spinal anesthesia). It is also used to maintain afterload in patients with aortic stenosis whose coronary perfusion is compromised by a decline in systemic vascular resistance. Given intravenously, phenylephrine has a rapid onset and relatively short duration of action (5 to 10 minutes). It may be given as a bolus of 40 to 100 μg or as an infusion starting at a rate of 10 to 20 μg/min. Larger doses, up to 1 mg, slow supraventricular tachycardia through reflex action. Phenylephrine is also a mydriatic and nasal decongestant. Applied topically, alone or in combination with local anesthetics, phenylephrine is used to prepare the nares for nasotracheal intubation.
α2-Adrenergic Agonists
α2-Agonists are assuming greater importance as anesthetic adjuvants and analgesics. Their primary effect is sympatholytic. They reduce peripheral norepinephrine release by stimulation of prejunctional inhibitory α2- receptors. Traditionally, they have been used as antihypertensive drugs, but applications based on their sedative, anxiolytic, and analgesic properties are becoming increasingly common.
Clonidine
Clonidine, the prototypical drug of this class, is a selective agonist for α2-adrenoreceptors. Its antihypertensive effects result from central and peripheral attenuation of sympathetic outflow. Clonidine withdrawal may precipitate a hypertensive crisis, so it should be continued throughout the perioperative period. A transdermal patch is available if a patient cannot take clonidine orally. If it is not continued perioperatively, arterial blood pressure should be monitored closely with ready ability to treat hypertension. Labetalol is used to treat clonidine withdrawal syndrome. Although experience with α(2)-agonists as a sole anesthetic is limited, these drugs can reduce the requirements for other intravenous or inhaled anesthetics as part of a general or regional anesthetic technique. (5) The results of a 2003 meta-analysis imply that perioperative use of clonidine and the other α2-agonists dexmedetomidine and mivazerol also decreased myocardial infarction and perioperative mortality rates in patients who had vascular surgery.6 However, a more recent (2014) large randomized trial of perioperative clonidine did not show a reduction in death or nonfatal myocardial infarction within 30 days of noncardiac surgery. (7)
In addition to their use in the operative setting, α2-agonists provide effective analgesia for acute and chronic pain, particularly as adjuncts to local anesthetics and opioids. Epidural clonidine is indicated for the treatment of intractable pain, which is the basis for approval of parenteral clonidine in the United States as an orphan drug. Clonidine also is used to treat patients with reflex sympathetic dystrophy and other neuropathic pain syndromes.
Dexmedetomidine
Like clonidine, dexmedetomidine is highly selective for the α(2)-receptors. Its half-life of 2.3 hours and distribution half-life of less than 5 minutes make its clinical effect quite short. Unlike clonidine, dexmedetomidine is available as an intravenous solution in the United States. The usual dosing is an infusion of 0.3 to 0.7 μg/kg/h either with or without a 1-μg/kg initial dose given over 10 minutes. In healthy volunteers, dexmedetomidine increases sedation, analgesia, and amnesia; it decreases heart rate, cardiac output, and circulating catecholamines in a dose-dependent fashion. The inhaled anestheticsparing, sedative, and analgesic effects demonstrated in preclinical and volunteer studies have been borne out in clinical practice. The relatively minor impact of α(2)- induced sedation on respiratory function combined with the short duration of action of dexmedetomidine has led to its use for awake fiberoptic endotracheal intubation. (8) Dexmedetomidine infusions for the perioperative management of obese patients with obstructive sleep apnea minimized the need for narcotics while providing adequate analgesia. (9)
4. Β2-ADRENERGIC RECEPTOR AGONISTS
β2-Agonists are used to treat reactive airway disease. With large doses the β2-receptor selectivity can be lost, and severe side effects related to β1-adrenergic stimulation are possible. Commonly used agonists include metaproterenol (Alupent, Metaprel), terbutaline (Brethine, Bricanyl), and albuterol (Proventil, Ventolin). β2-Agonists are also used to arrest premature labor. Ritodrine (Yutopar) has been marketed for this purpose. Unfortunately, β1-adrenergic adverse effects are common, particularly when the drug is given intravenously.
5. Α-ADRENERGIC RECEPTOR ANTAGONISTS
α1-Antagonists have long been used as antihypertensive drugs, but their side effects, which include marked orthostatic hypotension and fluid retention, have made them less popular as other medications for controlling arterial blood pressure with more attractive side effect profiles have become available.
Phenoxybenzamine
Phenoxybenzamine (Dibenzyline) is the prototypical α1-adrenergic antagonist (though it also has α(2)-antagonist effects). Because it irreversibly binds α1-receptors, new receptors must be synthesized before complete recovery. Phenoxybenzamine decreases peripheral resistance and increases cardiac output. Its primary adverse effect is orthostatic hypotension that can lead to syncope with rapid changes when patients move from the supine to standing positions. Nasal stuffiness is another effect. Phenoxybenzamine is most commonly used in the treatment of pheochromocytomas. It establishes a “chemical sympathectomy” preoperatively that makes arterial blood pressure less labile during surgical resection of these catecholamine-secreting tumors. When exogenous sympathomimetics are given after α1-block their vasoconstrictive effects are inhibited. Despite its irreversible binding to the receptor, the recommended treatment for a phenoxybenzamine overdose is an infusion of norepinephrine because some receptors remain free of the drug; vasopressin may also be effective in this setting.
Prazosin
Prazosin (Minipress) is a potent selective α1-blocker that antagonizes the vasoconstrictor effects of norepinephrine and epinephrine. Orthostatic hypotension is a major problem with prazosin. Unlike other antihypertensive drugs, prazosin improves lipid profiles by lowering low-density lipid levels and raising the level of high-density lipids. The usual starting dose of prazosin is 0.5 to 1 mg given at bedtime because of the risk of orthostatic hypotension. Doxazosin (Cardura) and terazosin (Hytrin) have pharmacologic effects similar to those of prazosin but have longerpharmokinetic half-lives. Because of the high cost of phenoxybenzamine, these agents are being used with greater frequency for the preoperative preparation of patients with pheochromocytomas. However, because these agents provide competitive antagonism instead of permanent binding to the α-receptors, modest intraoperative hypertensive episodes seem to be more common in these patients than in those who received phenoxybenzamine. Agents such as tamsulosin (Flomax) show selectivity for the α1A-receptor subtype and are effective in the treatment of benign prostatic hypertrophy without the hypotensive effects seen when the nonselective α1-blockers are used to treat this condition.
Yohimbine
α(2)-Antagonists such as yohimbine increase the release of norepinephrine, but they have found little clinical utility in anesthesia.
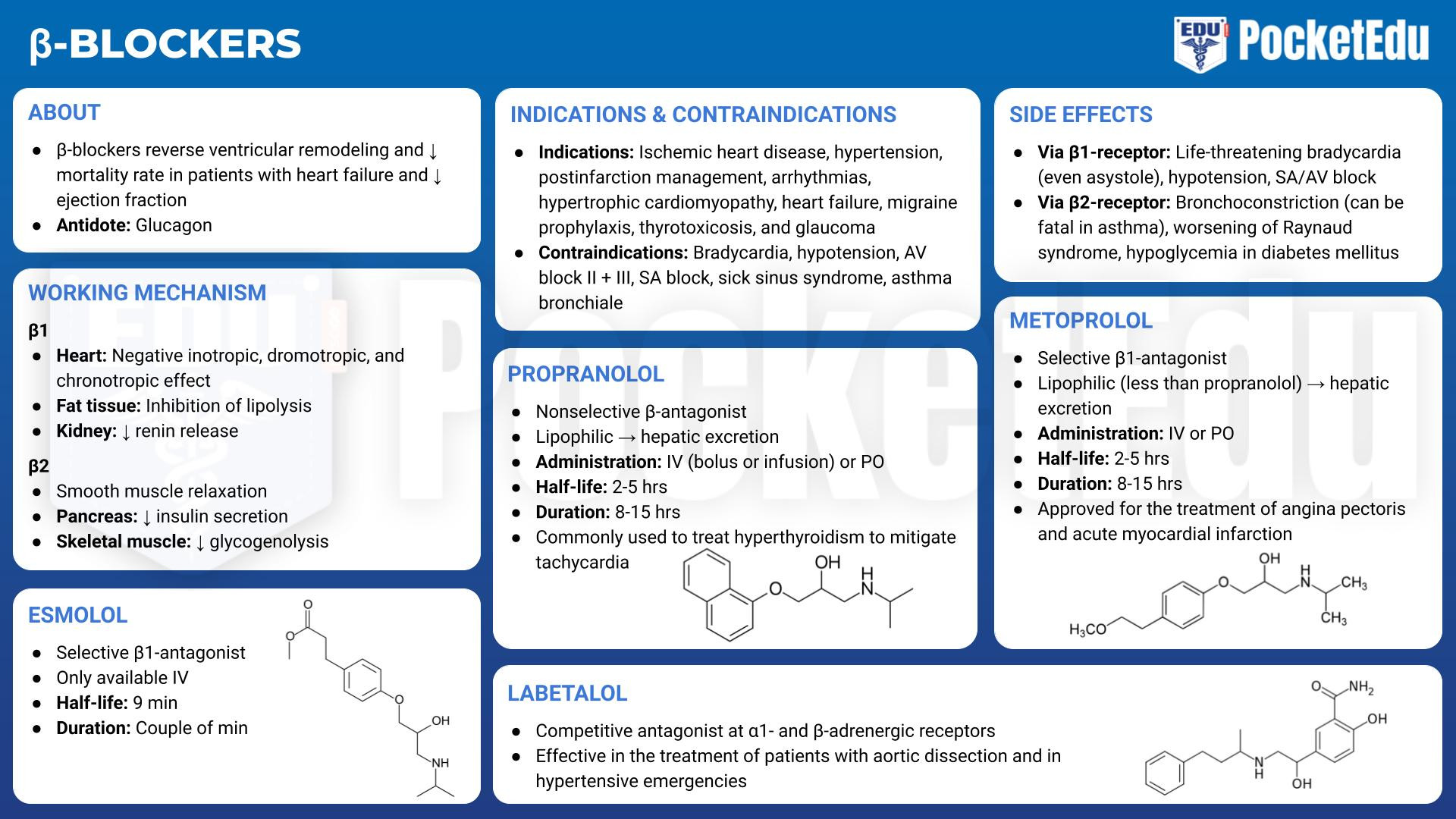
6. Β-ADRENERGIC ANTAGONISTS
β-Adrenergic antagonists (i.e., β-blockers) are frequently taken by patients about to undergo surgery. Clinical indications for β-adrenergic block include ischemic heart disease, postinfarction management, arrhythmias, hypertrophic cardiomyopathy, hypertension, heart failure, migraine prophylaxis, thyrotoxicosis, and glaucoma. In patients with heart failure and reduced ejection fraction, β-blocker therapy has been shown to reverse ventricular remodeling and reduce mortality rate. (10) In the 1990s, a study by the Perioperative Ischemia Research Group demonstrated the value of initiating β-block perioperatively in patients at risk for coronary artery disease. (11) Study subjects given perioperative β-blockers had a markedly reduced all-cause 2-year mortality rate (68% survival rate in placebo group vs. 83% in atenolol-treated group). The presumed mechanism for this improved survival rate was a diminution of the surgical stress response by the β-blockers. These and other confirmatory findings led to tremendous political and administrative pressure to increase the use of β-blockers perioperatively. Subsequent studies, however, have questioned the value of perioperative β-block, including a large study of oral metoprolol started on the day of surgery and continuing for 30 days (POISE trial), which demonstrated increased mortality rate in the β-blocker group. (12) A systematic review on perioperative β-block from the American College of Cardiology/American Heart Association (ACC/AHA) states that although perioperative continuation of β-block started 1 day or less before noncardiac surgery in high-risk patients prevents nonfatal myocardial infarctions, it increases the rate of death, hypotension, bradycardia, and stroke. In addition, there is insufficient data regarding continuation of β-block started 2 days or more before noncardiac surgery. (13) The 2014 ACC/ AHA Guideline on Perioperative Cardiovascular Evaluation and Management of Patients Undergoing Noncardiac Surgery recommends that patients on chronic β-blocker therapy continue this therapy in the perioperative period, but β-blocker therapy should not be started on the day of surgery. (14)
The most widely used β-adrenergic blockers in anesthetic practice are propranolol, metoprolol, labetalol, and esmolol because they are available as intravenous formulations and have well-characterized effects. The most important differences among these blockers are tied to cardioselectivity and duration of action. Nonselective β-blockers act at the β1- and β2-receptors. Cardioselective β-blockers have stronger affinity for β1-adrenergic receptors than for β2-adrenergic receptors. With β1-receptor selective block, velocity of atrioventricular conduction, heart rate, and cardiac contractility decrease. The release of renin by the juxtaglomerular apparatus and lipolysis at adipocytes also decrease. With larger doses, the relative selectivity for β1-receptors is lost and β2- receptors are also blocked, with the potential for bronchoconstriction, peripheral vasoconstriction, and decreased glycogenolysis.
Adverse Effects of β-Adrenergic block
Life-threatening bradycardia, even asystole, may occur with β-adrenergic block, and decreased contractility may precipitate heart failure in patients with compromised cardiac function. In patients with bronchospastic lung disease, β2-block may be fatal. Diabetes mellitus is a relative contraindication to the long-term use of β-adrenergic antagonists because warning signs of hypoglycemia (tachycardia and tremor) can be masked and because compensatory glycogenolysis is blunted. To avoid worsening of hypertension, use of β-blockers in patients with pheochromocytomas should be avoided unless α-receptors have already been blocked. Overdose of β-blocking drugs may be treated with atropine, but isoproterenol, dobutamine, or glucagon also may be required along with cardiac pacing to maintain an adequate rate of contraction.
Undesirable drug interactions are possible with β-blockers. The rate and contractility effects of verapamil are additive to those of β-blockers, so care must be taken when combining these drugs. Similarly, the combination of digoxin and β-blockers can have powerful effects on heart rate and conduction and should be used with special care.
Specific β-Adrenergic Blockers
Propranolol
Propranolol (Inderal, Ipran), the prototypical β-blocker, is a nonselective β-blocking drug. Because of its high lipid solubility, it is extensively metabolized in the liver, but metabolism varies greatly from patient to patient. Clearance of the drug can be affected by liver disease or altered hepatic blood flow. Propranolol is available in an intravenous form and was initially given as either a bolus or an infusion. Infusions of propranolol have largely been supplanted by the shorter-acting esmolol. For bolus administration, doses of 0.1 mg/kg may be given, but most practitioners initiate therapy with much smaller doses, typically 0.25 to 0.5 mg, and titrate to effect. Propranolol shifts the oxyhemoglobin dissociation curve to the right, which might account for its efficacy in vasospastic disorders. (15) Additionally, propranolol is commonly used in the treatment of hyperthyroidism to mitigate tachycardia that may result.
Metoprolol
Metoprolol (Lopressor), a cardioselective β-adrenergic blocker, is approved for the treatment of angina pectoris and acute myocardial infarction. No dosing adjustments are necessary in patients with liver failure. The usual oral dose is 100 to 200 mg/day taken once or twice daily for hypertension and twice daily for angina pectoris. Intravenous doses of 2.5 to 5 mg may be administered every 2 to 5 minutes up to a total dose of 15 mg, with titration to heart rate and blood pressure.
Labetalol
Labetalol (Trandate, Normodyne) acts as a competitive antagonist at the α1- and β-adrenergic receptors. Metabolized by the liver, its clearance is affected by hepatic perfusion. Labetalol may be given intravenously every 5 minutes in 5- to 10-mg doses or as an infusion of up to 2 mg/min. It can be effective in the treatment of patients with aortic dissection (16) and in hypertensive emergencies. Because vasodilation is not accompanied by tachycardia, labetalol has been given to cardiac patients postoperatively. It may be used to treat hypertension in pregnancy both on a long-term basis and in more acute situations. (17) Uterine blood flow is not affected, even with significant reductions in blood pressure (18).
Esmolol
Because it is hydrolyzed by blood-borne esterases, esmolol (Brevibloc) has a uniquely short half-life of 9 to 10 minutes, which makes it particularly useful in anesthetic practice. It can be used when β-block of short duration is desired or in critically ill patients in whom the adverse effects of bradycardia, heart failure, or hypotension may require rapid withdrawal of the drug. Esmolol is cardioselective, and the peak effects of a loading dose are seen within 5 to 10 minutes and diminish within 20 to 30 minutes. It may be given as a bolus of 0.5 mg/kg or as an infusion. When used to treat supraventricular tachycardia a bolus of 500 μg/kg is given over 1 minute, followed by an infusion of 50 μg/kg/min for 4 minutes. If the heart rate is not controlled, a repeat loading dose followed by a 4-minute infusion of 100 μg/kg/min is given. If needed, this sequence is repeated with the infusion increased in 50-μg/kg/min increments up to 300 μg/kg/min. Esmolol is safe and effective for the treatment of intraoperative and postoperative hypertension and tachycardia. If continuous use is required, it may be replaced by a longer-lasting cardioselective β-blocker such as metoprolol.
- PockedtEdu Anesthesia Infographics: Beta-blockers 6.6.
7. CHOLINERGIC PHARMACOLOGY
In contrast to the rich selection of drugs to manipulate adrenergic responses, there is a relative paucity of drugs that affect cholinergic transmission. A small number of direct cholinergic agents are used topically for the treatment of glaucoma or to restore gastrointestinal or urinary function. The classes of drugs with relevance to the anesthesia provider are the anticholinergic agents (muscarinic antagonists) and the anticholinesterases.
Muscarinic Antagonists
The muscarinic antagonists compete with neurally released ACh for access to muscarinic cholinoceptors and block ACh’s effects. The results are faster heart rate, sedation, and dry mouth. With the exception of the quaternary ammonium compounds that do not readily cross the blood-brain barrier and have few actions on the CNS, there is no significant specificity of action among these drugs; they block all muscarinic effects with equal efficacy, although there are some quantitative differences in effect. In the era of ether anesthetics, a muscarinic antagonist was added to anesthetic premedication to decrease secretions and to prevent harmful vagal reflexes. This addition is less important with modern inhaled anesthetics. Preoperative use of these drugs continues in some pediatric and otorhinolaryngologic cases or when fiberoptic intubation is planned.
Atropine with its tertiary structure can cross the bloodbrain barrier. Thus, large doses (1 to 2 mg) can affect the CNS. In contrast, because of the quaternary structure of the synthetic antimuscarinic drug glycopyrrolate (Robinul) it does not cross the blood-brain barrier. Glycopyrrolate has a longer duration of action than atropine and has largely replaced atropine for blocking the adverse muscarinic effects (bradycardia) of the anticholinesterase drugs that reverse neuromuscular block. Scopolamine also crosses the blood-brain barrier and can have profound CNS effects. The patch preparation of scopolamine is used prophylactically for postoperative nausea and vomiting, but it may be associated with adverse eye, bladder, skin, and psychological effects. The distortions of mentation (e.g., delusions or delirium) that can follow treatment with atropine or scopolamine are treated with physostigmine, an anticholinesterase that is able to cross the blood-brain barrier.
Cholinesterase Inhibitors
Anticholinesterase drugs impair the inactivation of ACh by the cholinesterase enzyme and sustain cholinergic agonism at nicotinic and muscarinic receptors. These drugs are used to reverse neuromuscular block and to treat myasthenia gravis. The most prominent side effect of these drugs is bradycardia. The commonly used cholinesterase inhibitors are physostigmine, neostigmine, pyridostigmine, and edrophonium. In addition to reversing the effects of neuromuscular blocking drugs by increasing the concentration of ACh at the neuromuscular junction, cholinesterase inhibitors stimulate intestinal function or are applied topically to the eye as a miotic. One topical drug (echothiophate iodide) irreversibly binds cholinesterase and can interfere with the metabolism of succinylcholine (as the anticholinesterases impair the function of the pseudocholinesterase enzyme as well).
8. QUESTIONS OF THE DAY
1. What are the cardiovascular, respiratory, endocrine, and metabolic effects of epinephrine? What are the expected cardiovascular effects of an intravenous epinephrine infusion as the dose increases?
2. How does the cardiovascular mechanism of action of phenylephrine differ from ephedrine?
3. What are the central nervous system, cardiovascular, and respiratory effects of dexmedetomidine infusion?
4. How does cardioselectivity and duration of action differ for the beta blockers available for intravenous use?
5. What are the most important differences in the side effect profile of the muscarinic antagonists atropine, glycopyrrolate, and scopolamine?


PRACTICAL
The gold-standard text in anesthesiology; used by everyone worldwide

BE INSPIRED
BONUS: INFOGRAPHICS
The best-selling practical book
enhanced with Flash Cards for tests
preparation.

UNDERSTAND
Make our own study scripts, insert images, YouTube or own videos, literature and never loose them.

READ OR LISTEN.
Learn according to your study style. Read or listen to the voiced-over material.