Learning objectives
- Difference between α- and β thalassemia
- Recognize thalassemia
- Management of thalassemia
Definition and mechanisms
- Thalassemia is an inherited group of hematological disorders
- Synthesis of alpha or beta globin chains is deficient, characterized by decreased hemoglobin production
- More frequently found in the Mediterranean, Central Africa, China, and South East Asian regions
- The majority of hemoglobin is HbA in normal adults, which has two α and two β chains, i.e. α2β2
α thalassemia
- α0 thalassemia results from the elimination of α1 and α2 genes and/or regulatory sequences
- α+ thalassemia minor results from a single gene deletion or point mutation that prevents normal α chain production
- The resulting anemia is mild and of little anesthetic significance
- Hemoglobin H disease is caused by the interaction of α0 and α+ determinants (loss of three genes)
- The clinical picture is of chronic hemolytic anemia with Hb levels of 8–10 g dL
- Stimulates erythropoietin production and results in excess production of large amounts of β-chains
- α thalassemia major” occurs when all four alpha globin genes are non-functioning
- Is almost uniformly fatal in utero without intervention
β thalassemia
- Is an autosomal recessive defect of the beta-globin chains of the hemoglobin A molecule
- The clinical presentation typically manifests at approximately 4 to 6 months of age
- A defect in one beta globin allele will result in beta thalassemia minor
- An effective carrier state as individuals are usually asymptomatic or present with anemia
- Hemoglobin levels 2-3 g/dL < normal for age
- A defect in both alleles results in beta thalassemia major (Cooley anemia)
- Results in profound anemia requiring lifelong blood transfusions
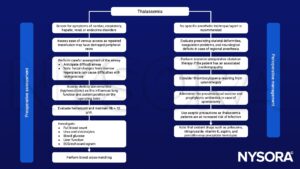
Signs and symptoms
| Intravascular hemolysis: | Perioperative anemia Jaundice, gallstones Splenic sequestration causes anemia and other hematological effects Free iron causes a vascular endothelial inflammatory response and its consequences, e.g. leading to systemic and Pulmonary hypertension (PH), cerebral ischemia, platelet activation, hypercoagulability |
| Extramedullary erythropoiesis: | Frontoparietal bossing, prominent maxilla and zygomatic arch, compression of neural structures in bony canal including the spinal cord, bony deformities e.g. thoracic cage, spinal column |
| Systemic effects of anemia: | High output cardiac failure, decreased oxygen delivery leading to poor growth |
| Multiple transfusions: | Hypervolemia, coagulation defects, immunosuppression, iron overload |
| Iron overload: | Cardiomyopathy, cardiac hypertrophy, myocarditis, pericarditis Liver failure and cirrhosis Renal tubular dysfunction, loss of concentrating renal function, and increased excretion of calcium, magnesium, and phosphate Endocrionpathy (diabetes, hypothyroidism, hypoparathyroidism, adrenal insufficiency) Immunosuppression: increased risk of infection |
Diagnosis
- Complete blood count
- Hemoglobin electrophoresis
- DNA-testing
- Metzer index, not a definitive test but in can suggest the possibility of thalassemia
Management
Suggested reading
- Pollard BJ, Kitchen, G. Handbook of Clinical Anaesthesia. Fourth Edition. CRC Press. 2018. 978-1-4987-6289-2.
- Thomas, C., Lumb, A.B., 2012. Physiology of haemoglobin. Continuing Education in Anaesthesia Critical Care & Pain 12, 251–256.
- Wilson, M., Forsyth, P., Whiteside, J., 2010. Haemoglobinopathy and sickle cell disease. Continuing Education in Anaesthesia Critical Care & Pain 10, 24–28.
We would love to hear from you. If you should detect any errors, email us customerservice@nysora.com